ABACUS(原子算筹)作为一款国产的电子结构软件,可用于对材料进行密度泛函理论(Density Functional Theory,简称 DFT)的第一性原理(first-principles)计算,其主要核心功能之一是电子自洽迭代计算 (Self Consistent Field,简称 SCF),在给定材料微观层面的晶胞和原子位置的条件下,我们可以通过 SCF 流程的计算获得电子结构的总能量、能级、电子波函数、电子密度等关键信息,这些信息可用于进一步计算得出材料体系的其它性质。然而,对于初学者来说,初次运行 ABACUS 难免不熟悉输入参数,容易出错,要熟练掌握好 SCF 计算要领也颇具难度。因此,本文将主要针对初学者,详细介绍(1)如何采用 ABACUS 设置合理的输入参数完成一个 SCF 计算,以及(2)如何对体系做收敛性测试,以便确定所有计算参数。这里收敛性测试包括在平面波(Plane Wave,简称 pw)基矢量下面对 ecut(电子动能截断值)进行测试,以及对布里渊区的 k 点个数进行测试来确定这些参数。
对密度泛函理论计算不太了解的读者,我们想先介绍密度泛函理论的三个特点。第一,只要给定原子坐标和种类,密度泛函理论就可以用于预测该体系的许多物理和力学等性质。第二,对于不同的原子坐标排列方式以及不同元素种类,密度泛函理论软件提供了一系列参数用于调节计算的精度和效率,只有将参数设定在某一些正确的范围之内,得到的结果才有意义。反之,如果参数过于粗糙,则得到的结果可能也不收敛,因此不具有价值。第三,密度泛函理论计算是一种虽然较为流行的但也相对昂贵的科学计算方法。笔记本电脑只能跑得动几个原子的小体系计算,真正数十个原子乃至更大的体系需要借助超算进行长时间的计算(几个小时甚至几天),而选择不同的参数可能会对最终运行时间有较大的影响。因此,基于以上三点认识,有必要更加细致地掌握密度泛函理论计算的具体参数,下面我们予以介绍。
根据布洛赫定理,周期性势场中的电子波函数可分解为平面波基组的线性组合:
其中:
-
倒格矢
$\mathbf{G}$ :由晶格实空间周期性导出,满足$\mathbf{G} \cdot \mathbf{R} = 2\pi n$ ($\mathbf{R}$为晶格矢量,$n$为整数) -
波矢
$\mathbf{k}$ :位于第一布里渊区(动量空间的基本周期单元),其取值范围由晶格对称性决定
正交性优势:平面波基函数
平面波基组理论上需要无限多个,但实际计算中引入 截断能($E_{\text{cut}}$) 以筛选动能较小的平面波:
其中
-
计算精度与效率平衡:
提高$E_{\text{cut}}$ 可提升精度(如石墨烯计算中常用 280 eV),但计算量随基组规模立方增长($N_{\text{PW}} \propto E_{\text{cut}}^{3/2}$)。 -
赝势类型的影响:
- 超软赝势(USPP):截断能需求最低(约 30-50 Ry)
- 模守恒赝势(NCPP):需更高截断能(如 100 Ry)
- 全电子计算:需极大截断能(>1000 Ry)
-
材料特性:
-
局域化程度:强局域化体系(如含 d/f 电子的过渡金属)需更高
$E_{\text{cut}}$ - 成键类型:离域化体系(如金属)可适当降低
-
局域化程度:强局域化体系(如含 d/f 电子的过渡金属)需更高
-
收敛性测试:通过逐步增加
$E_{\text{cut}}$ 直至总能量变化收敛(通常要求$\Delta E < 1,\text{meV/atom}$ ) -
参数示例:
- 石墨烯:$E_{\text{cut}}=280,\text{eV}$(超软赝势)
- 硅晶体:$E_{\text{cut}}=400,\text{eV}$(PAW 赝势)
| 优势 | 局限性 |
|---|---|
| 1. 正交归一性:无需计算重叠积分,哈密顿矩阵对角化高效 | 1. 芯电子描述困难:需极高截断能或赝势辅助 |
| 2. 参数系统性:仅需调节截断能即可控制精度,收敛性明确 | 2. 计算量随体系尺寸剧增(如超胞体积翻倍,计算时间增 8 倍) |
| 3. 非局域性:基组与原子位置无关,避免基组叠加误差 | 3. 强关联体系需结合 DFT+U 或 DMFT 等修正方法 |
总结:平面波基组通过能量截断和赝势技术实现了高效计算,其核心优势在于正交性与参数可控性,但在处理强局域化体系时需结合赝势或更高截断能策略。
赝势(Pseudopotential)是一种有效势场,用于简化量子力学多体问题的计算。其核心思想是:
- 冻结芯电子:将原子核与内层电子(芯电子)的复杂相互作用等效为一个平滑势场,仅显式处理外层价电子。
- 波函数平滑化:构造无节点的赝波函数,避免真实波函数在核附近的快速振荡,从而减少平面波基组的数量。
- 物理量守恒:在截断半径外,赝势的散射性质与真实势一致,保证能带、电荷密度等关键物理量的正确性。
-
特征:
- 要求赝波函数在截断半径外的模与真实波函数一致(电荷密度积分守恒)。
- 非局域性:不同角动量量子数($l$)对应不同势函数,适用于强局域化体系(如过渡金属)。
- 典型泛函:Hamann-Schlüter-Chiang (HSC) 势。
- 特征:
- 放宽模守恒条件,允许更平滑的赝波函数,显著减少平面波基组数量(计算效率提升 5-10 倍)。
- 需引入广义本征值问题,计算电荷密度时需补偿核区域信息。
- 典型泛函:Vanderbilt 超软势。
- 特征:
- 基于实验数据(如能带结构、光学谱)拟合势参数,适用于特定材料体系(如半导体 Si、Ge)。
- 计算量小,但可移植性差。
- 定位:严格来说非传统赝势,但实现类似效果。
- 特征:
- 通过投影算符连接全电子波函数与平滑赝波函数,精度接近全电子计算。
- 适用于强关联体系(如磁性材料、表面催化)。
- 半导体能带:经验赝势法用于计算 Si、GaAs 的带隙与光学性质。
- 金属近自由电子模型:模守恒赝势解释金属中价电子的类自由电子行为。
- 催化反应:PAW 方法用于研究表面吸附与反应路径。
- 异质结界面:超软赝势计算界面电荷转移与能带对齐。
- 纳米 MOSFET:经验赝势结合周期性边界条件模拟 10 nm 级晶体管的量子输运。
- 量子点与量子线:模守恒赝势研究尺寸效应导致的能谷劈裂。
- 材料数据库:超软赝势的高效性支持百万原子体系的大规模筛选(如 Materials Project)。
| 场景 | 推荐类型 | 原因 |
|---|---|---|
| 高精度能带计算 | PAW 或 NCPP | 电荷密度与全电子结果一致 |
| 大规模体系(>1000 原子) | USPP | 平面波截断能低(~30-50 Ry) |
| 半导体光学性质 | 经验赝势 | 参数化拟合与实验光谱高度匹配 |
| 磁性/强关联材料 | PAW | 精确处理局域 d/f 电子 |
赝势的引入是平衡计算精度与效率的关键策略,主要解决以下核心问题:
- 真实原子中,内层(芯)电子的波函数在原子核附近呈现剧烈振荡(如 d/f 轨道),需高能平面波基组(截断能 >1000 eV)才能准确描述。
- 赝势作用:通过冻结芯电子并构造平滑的赝波函数,消除快速振荡分量,将截断能降至 30-500 eV,使平面波基组规模减少 2-3 个数量级。
- 全电子计算(不冻结芯电子)需处理大量高能平面波,计算量随原子数呈立方增长,难以处理>100 原子的体系。
- 赝势方案:超软赝势(USPP)和 PAW 方法可将单原子平面波数量压缩至 50-100 个,显著提升大体系(如纳米器件、催化剂表面)的模拟能力。
- 化学反应和材料性质主要由价电子决定,芯电子贡献可近似处理(如相对论效应通过赝势参数化)。
- 赝势优势:在保留价电子散射特性的同时,忽略芯电子动态细节,确保能带、电荷密度等关键物理量的计算精度。
在密度泛函理论(DFT)框架中,赝势贯穿以下核心环节:
-
有效势场构造:赝势替代原子核与芯电子的真实势场,生成有效势能项:
$$ V_{\text{eff}}(\mathbf{r}) = V_{\text{ext}} + V_{\text{Hartree}} + V_{\text{xc}} + V_{\text{pseudo}} $$
其中$V_{\text{pseudo}}$ 包含赝势对价电子的等效作用。
- 赝势决定了平面波基组的截断能(如 USPP 截断能为 30-50 Ry,PAW 为 40-80 Ry),直接影响哈密顿矩阵的维度和计算耗时。
- POTCAR 文件:在 VASP、Quantum ESPRESSO 等软件中,赝势以 POTCAR 文件形式输入,包含不同元素的赝势参数(如 PAW_PBE 目录下的原子势文件)。
- 泛函兼容性:赝势需与交换关联泛函(如 PBE、LDA)匹配,例如 PAW-PBE 赝势需搭配 PBE 泛函使用以避免误差。
- 磁性材料:PAW 赝势通过投影算符精确描述 d/f 电子的局域磁矩,优于超软赝势。
- 强关联体系:DFT+U 方法需基于 PAW 赝势引入 Hubbard 参数修正,处理电子强关联效应。
总结:赝势是 DFT 计算中连接物理模型与数值实现的桥梁,通过简化芯电子处理、降低截断能需求,使平面波方法能够高效应用于材料模拟。其在 Kohn-Sham 势场构建、软件输入参数设计及特殊体系修正中均发挥核心作用。
布里渊区是倒易空间的基本单元,与实空间晶格的周期性形成傅里叶变换对:
-
倒格矢定义:若实空间晶格基矢为
$\bold{a}_1, \bold{a}_2, \bold{a}_3$ ,倒格矢$\bold{G}$ 满足$\bold{G} \cdot \bold{R} = 2\pi n$ ($\bold{R}$为晶格矢量,$n$为整数)。 -
第一布里渊区:由最邻近倒格矢的中垂面围成的最小闭合区域。例如:
- 简单立方晶格:立方体;
- 面心立方晶格(如铜):截角八面体(14 个面,对称性最高)。
-
物理意义:电子波函数
$\psi_{\bold{k}}(\bold{r})$ 的波矢$\bold{k}$ 被限制在第一布里渊区内,其能带结构在此区域内周期性重复。
-
正格子与倒格子
-
正格子:真实空间中晶体原子排列的周期性结构,由基矢
$\mathbf{a}_1, \mathbf{a}_2, \mathbf{a}_3$ 定义。 -
倒格子:倒易空间中的抽象点阵,基矢
$\mathbf{b}_1, \mathbf{b}_2, \mathbf{b}_3$ 满足: $$ \mathbf{a}i \cdot \mathbf{b}j = 2\pi \delta{ij} $$ 其中 $\delta{ij}$ 为克罗内克函数,当 i=j 时为 1,否则为 0。倒格子基矢的数学表达式为: $$ \mathbf{b}_i = 2\pi \frac{\mathbf{a}_j \times \mathbf{a}_k}{\mathbf{a}_i \cdot (\mathbf{a}_j \times \mathbf{a}_k)} $$ 其中$i, j, k$ 为循环排列。
-
正格子:真实空间中晶体原子排列的周期性结构,由基矢
-
倒易空间的物理意义
- 倒易空间中的点(倒格矢
$\mathbf{G}$ )对应正格子中的晶面族,其方向为晶面法线方向,大小为晶面间距的倒数乘以$2\pi$ 。例如,倒格矢$\mathbf{G} = h\mathbf{b}_1 + k\mathbf{b}_2 + l\mathbf{b}_3$ 对应正格子中密勒指数为$(hkl)$ 的晶面族。
- 倒易空间中的点(倒格矢
-
几何定义
在倒易空间中,以某个倒格点(通常取原点)为中心,绘制所有相邻倒格矢的垂直平分面,这些平面包围的最小闭合区域即为第一布里渊区。-
垂直平分面方程:对每个倒格矢
$\mathbf{G}$ ,其垂直平分面方程为: $$ \mathbf{k} \cdot \mathbf{G} = \frac{1}{2}|\mathbf{G}|^2 $$ 其中$\mathbf{k}$ 为倒空间中的波矢。
-
垂直平分面方程:对每个倒格矢
-
示例:简单立方晶格
-
正格子基矢:
$\mathbf{a}_1 = (a, 0, 0)$ ,$\mathbf{a}_2 = (0, a, 0)$ ,$\mathbf{a}_3 = (0, 0, a)$ -
倒格子基矢:
$\mathbf{b}_1 = (2\pi/a, 0, 0)$ ,$\mathbf{b}_2 = (0, 2\pi/a, 0)$ ,$\mathbf{b}_3 = (0, 0, 2\pi/a)$ -
第一布里渊区:由相邻倒格矢的垂直平分面围成立方体,边长为
$2\pi/a$ 。
-
正格子基矢:
-
高对称性点的意义
- Γ点(原点):对应波矢
$\mathbf{k} = 0$ - X 点:立方体边界的中心点
- L 点:面心立方晶格的特殊对称点
- Γ点(原点):对应波矢
-
电子波的布拉格反射
当电子波矢$\mathbf{k}$ 到达布里渊区边界时,满足布拉格反射条件: $$ 2\mathbf{k} \cdot \mathbf{G} = |\mathbf{G}|^2 $$ 导致电子波被晶格强烈散射,形成驻波和能隙(禁带)。 -
动量空间的周期性
第一布里渊区外的波矢可通过平移倒格矢$\mathbf{G}$ 映射回第一区内: $$ \mathbf{k}' = \mathbf{k} + \mathbf{G} $$ 此时$\mathbf{k}'$ 与$\mathbf{k}$ 描述相同的物理状态。 -
实际应用
- 能带计算:通过第一布里渊区内的 k 点采样计算电子能量分布
-
材料性质分析:布里渊区形状影响材料的导电性、光学特性等
(例如面心立方晶体的布里渊区为截角八面体)
第一布里渊区是倒易空间中的最小重复单元:
- 通过相邻倒格矢的垂直平分面构造
- 边界对应晶格对电子波的最大散射条件
- 简化了晶体中量子态的描述,是能带理论和衍射分析的核心工具
在 DFT 中,电子性质的计算需对布里渊区积分:
$$ E = \frac{V_{\text{cell}}}{(2\pi)^3} \int_{\text{BZ}} f(\bold{k}) d\bold{k} $$
其中
- 对称性优化:利用晶格点群对称性,将全布里渊区积分约化为不可约布里渊区(IBZ),计算点数减少至 1/48(立方晶系)。
- 误差来源:k 点不足会导致能带、电荷密度等物理量失真,尤其对金属体系费米面附近的电子态敏感。
-
数学形式:均匀网格 k 点坐标为:
$$ \bold{k}{n_1,n_2,n_3} = \sum{i=1}^3 \frac{2n_i - N_i -1}{2N_i} \bold{G}_i $$
其中
$N_i$ 为各方向网格密度,$\bold{G}_i$ 为倒格子基矢。 -
适用性:
- 金属:费米面附近能带剧烈变化,需高密度网格(如 12×12×12);
- 表面/低维体系:非周期方向(如薄膜 z 方向)仅需 1 个 k 点。
-
物理思想:通过对称性分析选择高权重 k 点,例如 Baldereschi 点
$(\pi/a)(1/2,1/2,1/2)$ ,其函数特性满足奇对称性与正交性,减少积分误差。 -
适用性:
- 半导体/绝缘体:能带平缓,少量特殊点即可逼近积分结果;
- 强关联体系:结合对称性权重修正局域电子行为。
| 材料类型 | 典型网格 | 物理依据 |
|---|---|---|
| 金属 | 8×8×8 至 16×16×16 | 费米面处电子态密度陡变,需密集采样 |
| 半导体 | 4×4×4 至 6×6×6 | 能带结构平缓,中等密度保证收敛 |
| 二维材料 | 12×12×1 | 平面内电子离域性强,垂直方向局域化 |
收敛性测试物理判据:
-
总能量变化:相邻网格总能量差
$\Delta E < 1,\text{meV/atom}$ ; - 态密度收敛:金属需检查费米能级附近态密度是否平滑。
高对称点是布里渊区内对称性最高的位置,通过连接这些点的路径可以反映整个布里渊区的能带特征,从而大幅减少计算量:
- 能带路径选择:计算能带时,只需沿高对称点连线(如Γ→X→M→Γ)采样,即可完整描述电子能量分布。
- 对称性优化:利用晶格点群对称性,将全布里渊区积分压缩到不可约布里渊区(IBZ),计算点数可减少至 1/48(如立方晶系)。
高对称点的位置和能量特征直接关联晶体的对称性和电子行为:
- 能级简并分析:在高对称点(如Γ点、X 点),电子能级常因对称操作(如旋转、反射)发生简并,导致能带交叉或能隙形成。
- 材料分类:通过能带在高对称点的特征,可区分导体、半导体和绝缘体。例如,导体的费米面常穿过能带,而半导体的带隙出现在高对称点之间。
高对称点为材料的光学、电学和热学性质研究提供理论依据:
- 光谱分析:拉曼光谱、X 射线衍射等实验中,高对称点的振动模式或衍射峰位置与对称性密切相关。
- 功能材料设计:在超分子化学中,高对称性结构(如正八面体超分子笼)可通过对称点设计实现高效催化或分子识别功能。
在密度泛函理论(DFT)等计算中,高对称点是 K 点采样的关键参考:
- Monkhorst-Pack 方法:通过均匀网格在高对称区域密集采样(如金属需 16×16×16 网格以捕捉费米面变化)。
- Chadi-Cohen 特殊点法:针对绝缘体/半导体,选取少数高对称点(如 Baldereschi 点)即可逼近积分结果。
- 拓扑量子化学:通过分解高对称点的能带表示,判断材料的拓扑性质(如表面电子态分析)。
- 非平衡态模拟:超快激光/强场作用下,高对称点附近的电子激发路径对材料瞬态响应至关重要。
总结
高对称性点是连接晶体对称性、电子行为与材料功能的桥梁,其应用范围从基础能带计算扩展到拓扑材料、催化设计和量子计算等前沿领域,是凝聚态物理和材料科学研究的核心概念。
核心方程: $$ \psi_{\mathbf{k}}(\mathbf{r}) = \sum_{i} c_i \chi_i(\mathbf{r} - \mathbf{R}_i) $$
-
原子轨道基底:$\chi_i$需满足正交归一性,常用类型包括:
- 数值原子轨道 (NAO):通过径向函数截断实现局域化(如 DZP 基组截断半径 5-7 Å)
- Slater 型轨道 (STO):精确描述原子轨道但积分计算复杂,需用 Gauss 函数拟合(如 STO-3G 基组)
- Gauss 型轨道 (GTO):数学处理高效,适用于大体系(如 6-31G *基组)
-
周期性扩展:固体计算中引入 Bloch 相位因子,形成周期匹配的基函数: $$ \chi_{\mathbf{k},i}(\mathbf{r}) = e^{i\mathbf{k} \cdot \mathbf{R}_i} \chi_i(\mathbf{r}-\mathbf{R}_i) $$ 该形式在 QuantumATK 等软件中用于处理周期性边界条件
| 基组类型 | 优化方法 | 典型应用场景 |
|---|---|---|
| SZ | 单ζ轨道,截断半径优化(~5 Å) | 快速结构弛豫 |
| DZP | 双ζ+极化轨道,提升价电子描述 | 半导体能带计算 |
| TZDP | 三ζ+双极化,增加角动量量子数 | 精确反应能垒预测 |
| aug-cc | 添加弥散函数,增强弱相互作用描述 | 氢键/范德华体系 |
截断半径影响:增大半径提升基组完备性但增加计算量,需通过能量收敛测试确定最优值(如 Si 体系 DZP 基组截断半径 6 Å时能量误差<0.01 eV/atom)
- 稀疏矩阵存储:利用原子轨道局域性,仅存储非零矩阵元(稀疏度>90%时可节省 80%内存)
- 赝势兼容性:
- PAW 赝势:需匹配投影算符与基组(如 SG15 赝势+DZP 基组)
- 超软赝势:允许更小截断半径,但需处理额外重叠积分项
- 并行算法:基于 MPI+OpenMP 混合并行,实现 10^3 原子体系的高效计算(如 ABACUS 在 200 节点上达到 90%并行效率)
| 维度 | LCAO 方法 | 平面波方法 (PW) |
|---|---|---|
| 基组构成 | 原子中心局域轨道(如 DZP/TZDP) | 全局扩展平面波基底 |
| 截断控制 | 轨道截断半径(~6 Å) | 能量截断 Ecut(~500 eV) |
| 内存需求 | 稀疏矩阵存储(<10 GB/千原子) | 密集矩阵存储(>100 GB/千原子) |
| 计算精度 | 依赖基组质量(DZP 误差~0.1 eV) | 系统收敛性明确(Ecut控制) |
| 适用体系 | 表面/界面/非周期体系(局域性强) | 完美晶体/均匀电子气(周期性优) |
| 指标 | LCAO(DZP) | PW(Ecut=50 Ry) |
|---|---|---|
| 单步 SCF 时间 (s) | 120 | 450 |
| 内存占用 (GB) | 8.2 | 32.5 |
| 带隙误差 (eV) | 0.15 | 0.05 |
| 并行扩展性 | 线性至 512 核 | 超线性至 10^4 核 |
平面波-LCAO 耦合:
- 区域分割:近原子区用 LCAO 描述局域轨道,远场区用平面波处理离域电子
- 接口处理:通过投影算符实现基组间电荷密度传递(如 QuantumATK 的 Embedding 模块)
- 精度提升:对表面吸附体系,耦合方法可降低 30%计算量同时保持<0.02 eV 精度
- 基组优化:利用神经网络自动生成最优数值原子轨道(如 DeepH 模型 MAE<0.05 eV)
- 矩阵预测:通过图神经网络直接预测哈密顿矩阵,跳过 SCF 迭代(加速比>10x)
- QM/MM 耦合:LCAO 作为量子力学区核心,与分子力学力场无缝对接(如催化反应模拟)
- 非绝热动力学:结合 LCAO 基组与表面跳跃算法,实现光激发过程模拟(时间尺度>ps)
| 软件 | LCAO 特性 | PW 特性 | 混合计算能力 |
|---|---|---|---|
| ABACUS | 支持 DZP/TZDP 基组,截断半径自适应优化 | PAW 赝势,Ecut自动收敛测试 | LCAO+PW 电荷密度混合 |
| SIESTA | 局域轨道+超软赝势,内存优化 | 支持但非主流 | 支持 QM/MM 边界耦合 |
| VASP | 仅支持平面波基底 | PAW 赝势+投影缀加波,高精度 | 无 |
| QuantumATK | 自适应基组 (LCAO/PW 切换) | HSE06 杂化泛函兼容 | 实时基组混合 |
-
选择 LCAO:
- 体系>500 原子,需快速结构优化
- 表面吸附/缺陷等局域化体系
- 内存限制严格(<64 GB)
-
选择平面波:
- 小体系(<100 原子)高精度计算
- 均匀电子气/能带精细结构研究
- 需要严格收敛性控制的课题
-
混合方法:
- 异质界面电子结构
- 催化反应过渡态搜索
- 光电器件载流子迁移率计算
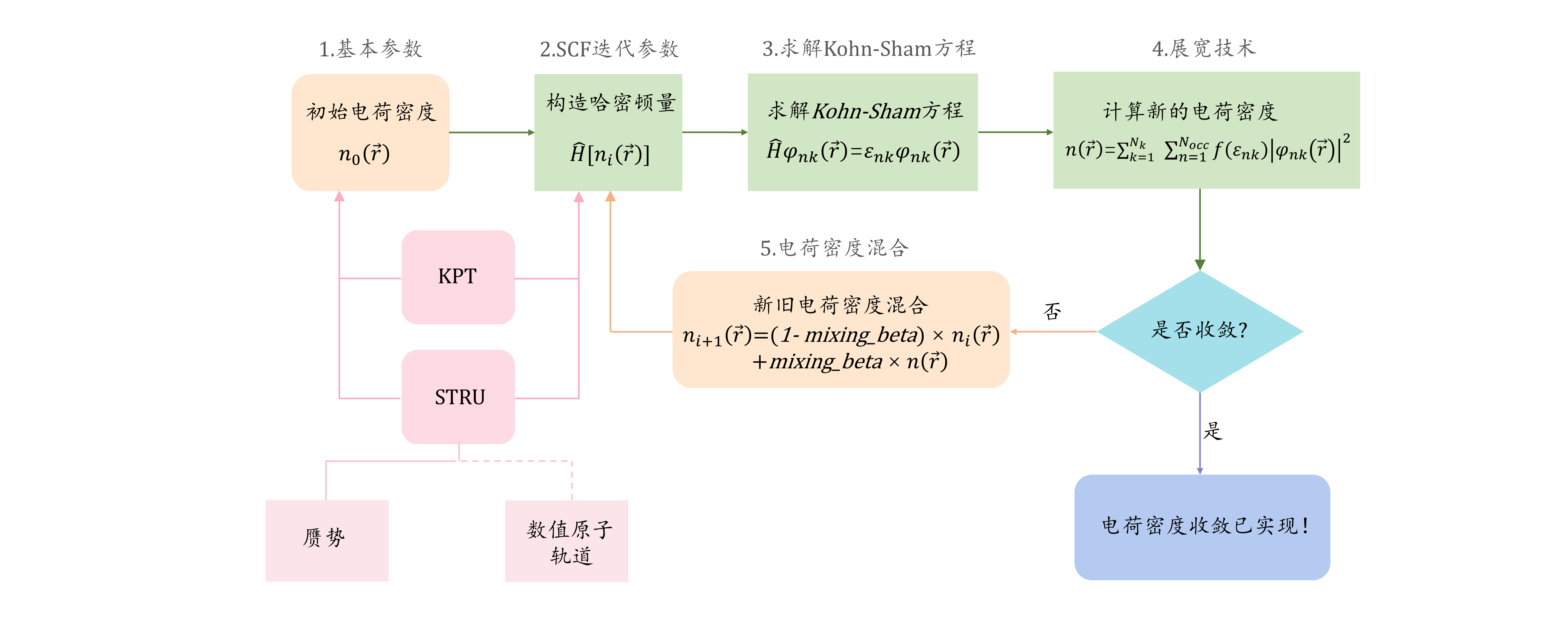
图 1:控制 SCF 的主要输入文件有三个。
KPT和STRU文件分别存储了布里渊区的信息和晶体结构的信息。此外INPUT文件主要存储了 5 部分信息,对应于下一章的小节序号,可以对应查看。$n_0(\vec{r})$表示体系运行开始时的初猜电荷密度,$\widehat{H}$表示电子系统的哈密顿量,$\varphi_{nk}(\vec{r})$表示波函数,其中$n$ 代表不同的能级,$k$代表体系的第一布里渊区里不同的采样点。$\varepsilon_{nk}$表示特征值,$N_{k}$代表$k$点的个数,$N_{occ}$表示被电子占据的能级个数,$f(\varepsilon_{nk})$表示电子的费米-狄拉克分布函数,$n(\vec{r})$表示由波函数算出来的电荷密度,$n_i(\vec{r})$表示迭代过程中第$i$步的电荷密度而$n_{i+1}(\vec{r})$表示第$i+1$步的电荷密度。$mixing_beta$表示混合新电荷密度的比例(取值范围为 0~1)。ABACUS 支持平面波和数值原子轨道双基组,如果使用平面波基矢量,则数值原子轨道信息可以在STRU里不提供。
使用 ABACUS 中进行密度泛函理论或者第一性原理分子动力学计算时,一般需要 STRU、KPT、INPUT 三个最基本的文件作为 ABACUS 的基本输入文件。STRU 文件提供元素、晶格、原子的基本结构信息;KPT 文件提供周期性边界条件下布里渊区采样的网格设置。INPUT 文件主要提供计算所需的各种设置参数,在图 1 里我们分成了五部分且将在下面详细介绍。
STRU 文件提供元素种类、原子质量(SCF 中不会用到)、赝势文件(如果做密度泛函理论计算)、数值原子轨道(如果采用该基组做计算)、晶格矢量、原子位置、原子位置是否可移动等基本结构相关的信息。其中,晶格是晶体中原子有序排列的具体形式,我们可以选取一组晶格矢量来描述晶格。晶格确定后,原子的相对位置也可以通过选取不同坐标系(例如 Cartesian 直角坐标系或者 Direct 晶格坐标系)确定下来。
例如,以下是一个金刚石硅的结构文件(8 个硅原子,周期性边界条件)
ATOMIC_SPECIES
Si 28.085 Si_ONCV_PBE-1.0.upf
NUMERICAL_ORBITAL
Si_gga_8au_60Ry_2s2p1d.orb
LATTICE_CONSTANT
1.8897261258369282
LATTICE_VECTORS
5.4307000000 0.0000000000 0.0000000000
0.0000000000 5.4307000000 0.0000000000
0.0000000000 0.0000000000 5.4307000000
ATOMIC_POSITIONS
Direct
Si
0.0000000000
8
0.0000000000 0.0000000000 0.0000000000 1 1 1 mag 0.0
0.0000000000 0.5000000000 0.5000000000 1 1 1 mag 0.0
0.5000000000 0.0000000000 0.5000000000 1 1 1 mag 0.0
0.5000000000 0.5000000000 0.0000000000 1 1 1 mag 0.0
0.7500000000 0.7500000000 0.2500000000 1 1 1 mag 0.0
0.7500000000 0.2500000000 0.7500000000 1 1 1 mag 0.0
0.2500000000 0.7500000000 0.7500000000 1 1 1 mag 0.0
0.2500000000 0.2500000000 0.2500000000 1 1 1 mag 0.0- 第 2 行 "元素 原子质量 赝势" 中的 "原子质量" 在进行 DFT 计算时必须写,哪怕在 SCF 中不会用到。那么什么时候会用到呢?比如在进行分子动力学(Molecular Dynamics,简称 MD,在 INPUT 的 calculation 参数里设置)计算时,涉及运动的积分方程时用的是牛顿的第二定律$F=ma$,此时会使用原子的质量。另外注意,此行需要提供所使用的 "赝势" 文件名称。
- 第 5 行提供 LCAO 基组所需的 "某种元素的数值原子轨道" 文件名称,而如果采用 pw 基组计算,不需要写第 4~5 行内容。
- 第 8 行代表晶格整体缩放的一个长度,注意1 Angstrom=1.8897261258369282 bohr,这里写 1.8 开头的小数意味着接下来的 LATTICE_VECTORS 部分可以写以 Angstrom 为单位的晶格。
- 第 11~13 行表示$x,y,z$方向分别对应的三条晶格矢量。
- 第 16 行 "Direct" 表示给出原子位置的方法是分数坐标(或者称为晶格坐标),常用的还有Cartesian(笛卡尔坐标方法),就是直角坐标系下的坐标。
- 第 17 行是元素种类的名称。
- 第 18 行设置原子初始磁矩,如果
INPUT里的nspin参数设为 1,则不考虑磁性,这个参数不起作用。 - 第 19 行表示体系的硅原子个数。
- 第 20~27 行给出所有 8 个硅原子的坐标;前三个数是坐标,之后的 (1 1 1) 三个数代表允许该原子在对应的$x,y,z$对应方向上移动,相反,(0 0 0) 表示不允许在对应的方向上移动;同样,当考虑磁性计算的时候,"mag 0.0" 指定每个原子的初始磁矩,若设置此参数,则第 18 行的值将被覆盖。
关于赝势,还值得多提一些。平面波基组计算涉及采用描述电子和离子吸引作用的赝势,例如模守恒赝势或者超软赝势。需要注意的是,如果赝势换了,则相应的 ecut 能量截断值也需要重新测试收敛性。而基于局域原子轨道 (Linear Combination of Atomic Orbitals,简称 LCAO) 基组的计算则涉及赝势和数值原子轨道(Numerical Atomic Orbitals,简称 NAO),需要使用者准备好相应文件。这里提到的赝势是一种描述核外电子和离子之间相互作用的近似方法,数值原子轨道是通过数值方法构建的描述电子波函数和电子密度等性质的基函数。需要注意的是,LCAO 计算所采用的数值原子轨道要和对应的赝势匹配,即数值原子轨道是从给定赝势生成出来的,赝势如果改变了,相应的数值原子轨道也要重新生成,才能保证较好的精度。一般来说数值原子轨道的截断半径越长或轨道越多,则基矢量越完备,计算精度越高,但同时计算量也越大。
KPT 文件提供周期性边界条件下布里渊区$k$点采样的网格设置
K_POINTS
0
Gamma
k k k 0 0 0- 第 2 行表示$k$点的总数,如果设置成 "0" 代表$k$点是自动生成的
- 如果第二行是 0,则第 3 行 "Gamma" (Γ-centered Monkhorst-Pack method) 是选择以 Gamma 点为中心的 Monkhorst-Pack 方法划分布里渊区网格,此外还可以使用 "mp" 方法,即最常用的 Monkhorst-Pack 方法。
- 第 4 行的前三个整数代表网格沿着每个方向划分成几份,后三个数代表网格的平移量,0 0 0 即代表不平移。
- 计算立方形晶格时,$k$点各方向应取相同个数。在本次计算中,我们会将$k$的各方向从 2 取到 8,测试不同$k$点下体系能量的收敛情况。
- 在后文我们将介绍如何进行$k$点的收敛性测试,当选定一个$k$点后,若体系在某方向扩胞$n$次,该方向$k$点个数大致可以缩小到原来的$\frac{1}{n}$。对于 N 原子以上的立方大体系,可通过在
INPUT文件中设置参数gamma_only取 1,即只使用 Gamma 点计算,这个时候计算所需内存会显著下降,计算效率也会有提升。 - 此外,$k$点还有所谓的
line mode,或者离散的$k$点模式,若有兴趣的读者可以查看KPT的介绍文档(http://abacus.deepmodeling.com/en/latest/advanced/input_files/kpt.html)。
我们以金刚石结构硅原子体系(8 个原子)作为示例,进行电子自洽迭代计算 (Self Consistent Field,SCF)。接下来让我们需要准备一个 INPUT 文件,我们把一些计算的关键参数分成如下 5 个部分。
-
suffix:
suffix是用户可以自定义的后缀,运行 ABACUS 可执行程序之后,输入文件所在的文件夹里会生成一个包含大部分运行信息的OUT.suffix文件夹。例如,在这个例子里我们可设为 "Si",运行后就会产生一个OUT.Si文件夹。 -
calculation:设置本次计算类型,例如本次文档主要展示 "scf"(自洽电子结构计算)。scf((Self Consistent Field)、relax、cell-relax、md(Molecular Dynamics) 是较常用的四类计算。
- scf(自洽电子结构计算)是一种用于求解电子基态电子密度的自洽迭代计算方法,通过迭代来持续更新体系的电子密度,以达到收敛条件。通过电子自洽迭代,计算是否达到收敛由参数
scf_thr决定。 - relax(结构弛豫计算)是通过调整体系中原子的位置来达到系统最稳定的状态的计算方法,每一次 relax 中都包含若干步的 scf 计算。在 relax 中,需要设置参数
force_thr_ev(eV/Angstrom),代表所有原子中最大受力的收敛阈值。而 relax 计算的收敛需要每一步离子迭代时 scf 中的scf_thr都满足,以及最后多步离子迭代后force_thr_ev也满足。 - cell-relax(结构弛豫计算)与 relax 不同的是,其晶胞参数,比如晶格常数、晶格形状也会发生变化。除
force_thr_ev外,还需要设置参数stress_thr(kbar),是晶胞感受到的应力的阈值。 - md(分子动力学)基于牛顿力学原理,通过数值积分模拟粒子的运动轨迹,根据原子间的势能计算相互作用力,因此势函数的选择比较关键,在 ABACUS 里可以选择 DFT 来计算相互作用力,也可以选择 DP 势函数。
- scf(自洽电子结构计算)是一种用于求解电子基态电子密度的自洽迭代计算方法,通过迭代来持续更新体系的电子密度,以达到收敛条件。通过电子自洽迭代,计算是否达到收敛由参数
-
symmetry:是否考虑对称性,有如下三个选项
-1 不进行对称性分析
0 仅考虑时间反演对称性
1 进行对称性分析
如果打开对称性(设置为 1),布里渊区$k$点可以根据对称性进行简化处理,若体系有对称性,则可以减少所需计算的$k$点。因为每个$k$点都会进行一次 Kohn-Sham 方程的求解,对称性分析后若 k 点减少则将提升计算效率。本次计算中开启对称性分析,设置 "1",关于对称性分析的测试还在进一步完善,如果计算结果奇怪,建议设置成 "0"之后再进行计算,比较结果是否一致。
-
pseudo_dir:计算中需要使用赝势来近似离子和电子相互作用势能,为计算提供赝势文件。pseudo_dir指定
STRU文件中赝势文件所在的目录。本次计算将赝势和轨道文件都与INPUT、STRU、KPT文件放在一起,因此填入 ".",表示处在当前文件夹中。ABACUS 支持的赝势文件——Si_ONCV_PBE-1.0.upf
"ONCV"代表模守恒赝势的种类,"PBE"是采用的交换关联泛函。
-
orbital_dir:本次计算将采取 pw(平面波轨道)基组(后文 basis_type 中设置)。若采用 LCAO 基组进行计算,则需要提供作为基组的轨道文件,在
STRU文件中指定轨道文件所在的目录。若选用 pw 基组,则不需要轨道文件,不需要填写此参数。常用的轨道有:DZP(Double-ζ valence orbitals plus SZ polarization,两条径向轨道和一条杂化轨道),TZDP(Triple-ζ valence orbitals plus DZ polarization,三条径向轨道和两条杂化轨道)。ABACUS 支持的轨道文件——Si_gga_7au_100Ry_2s2p1d.orb:
"gga"代表 GGA 泛函;"7au"是轨道半径,半径越大,计算结果会更准确,但花费时间也更久;"100Ry"是推荐的
ecutwfc值;"2s2p1d"表明采用了 DZP 轨道,轨道数目越多,即基矢量更完备,计算也会更准确。 -
basis_type:计算的基组(指描述电子波函数的基函数),在 ABACUS 中常用的有两种:
pw:平面波基组(由于基矢量更完备,pw 计算将更准确,同时计算时间也更长)
LCAO:局域原子轨道基组(没有 pw 准确,但效率高)
-
ecutwfc:平面波函数的能量截止(单位:Ry),在平面波基组里是很重要的一个参数,其大小决定着作为基矢的平面波函数的个数多少,而基矢的多少,也决定了计算精度的高低。
在本次计算选用的 LCAO 基组中,一般选取轨道文件上推荐的值,设置 "100" Ry 即可。
本次计算中选择 pw 基组。如前所述,pw 中不会用到轨道文件。
我们目前填写好的基本参数如下:
#Parameters (1.General)
suffix Si
calculation scf
symmetry 1
pseudo_dir .
orbital_dir .
basis_type pw
ecutwfc 100-
scf_nmax:针对每个离子构型,scf 电子迭代的最大次数为
scf_nmax,可设为"50"或"100" 次。半导体和绝缘体是较容易收敛的体系,一般 20 步 scf 以内即可达到收敛,对于金属体系或者费米面较为复杂的体系,有可能 50 步仍未收敛。注意,如果做 relax, cell-relax, 或者 md 的时候发现有某些 scf 迭代次数达到最大时仍未达到收敛条件,建议先把计算停下来弄清楚原因后再继续算,有可能是因为离子构型或者$k$点等参数的设置不合理引起的。 - scf_thr:代表两个相邻电子迭代步之间的电荷密度误差,也是 SCF 计算中判断是否收敛、完成计算的标准,对于 LCAO,一般设置为 "1e-7",可认为精度足够。对于 pw,建议设置 1e-8 或 1e-9 的精度。
#Parameters (2. SCF iterations)
scf_nmax 100
scf_thr 1e-8- nbands:计算的 Kohn-Sham 轨道数目,在本次计算中无磁性,参数
nspin取 1(默认值),程序目前采取0.5*max(1.2occupied_bands, occupied_bands + 10) 计算 nbands。 对于 Si,价电子数为 4,每个能级填充可以填充自旋向上和向下 2 个电子,金刚石结构中共有 8 个原子,则nbands=max(1.282,82+10)=26。 - ks_solver:在不同基组中展开哈密顿矩阵的对角化方法,对于 pw,可以选择
cg(Conjugate Gradient,默认方法),bpcg(还不是太稳定、测试中),dav(Davidson 算法);对于 LCAO,可以选择genelpa(默认值),scalapack_gvx(Scalable Linear Algebra PACKage)。如果选用 LCAO 基组,ks_solver可设置为"genelpa"。
#Parameters (3. Solve KS equation)
nbands 26
ks_solver cg- smearing_method:对于金属体系或者费米面附近较复杂的体系,电子自洽迭代方法往往不容易收敛,这个时候可以选择给电子提供一个光滑的占据函数,用来调节程序计算电荷密度和总电子数时候的电子占据函数,这对于费米面附近的电子态尤为重要。我们这里称为 "smearing method",或者称为展宽方法。具体来说,对于金属体系可以选择
mp(methfessel-paxton),mp2(2-nd methfessel-paxton) ,也可以使用gauss(也可以写作gaussian)。因为我们计算金刚石 Si 具有半导体性质,所以 smearing 方法基本不起作用,默认可以选择gauss。此外,非导体计算可以选择fixed,半导体和非导体也可以选择fd(Fermi-Dirac) 方法。 - smearing_sigma:给定展宽方法的能量范围(单位:Ry),默认是 0.015 Ry,我们按照通常情况给定 "0.01"。
#Parameters (4.Smearing)
smearing_method gauss
smearing_sigma 0.01- mixing_type:进行新旧电荷密度混合时选用的方法,可选 "plain"(简单电荷密度混合方法,如图 1 所示)、"pulay"、"broyden"),默认选择 "broyden"算法。
- mixing_beta:新旧电荷密度混合时,新电荷的比例,不同系统取值不同。能带大于 1 eV 的体系,设为 0.7;能带小于 1 eV 的金属和过渡金属,设为 0.2。这个参数越大,收敛得越快,但不收敛的风险也会变大,不过取值大小并不会改变基态能量的结果。对于 Si,因为其能带隙为 1.12 eV,本计算中设置此参数为 "0.7"。
- mixing_gg0:电子迭代过程中,可能出现混合电荷密度不收敛的情况,此时可以通过一种叫 Kerker Mixing 的方法来加快收敛。这个参数代表 Kerker 方法中调整电荷密度的尺度,本计算中设置此参数为 "0"。
#Parameters (5.Mixing)
mixing_type broyden
mixing_beta 0.7
mixing_gg0 0到此,我们完成了对主要计算参数的设置,此时集成了以上主要参数的 INPUT 文件如下:
#Parameters (1.General)
suffix Si
calculation scf
symmetry 1
pseudo_dir .
orbital_dir .
basis_type pw
ecutwfc 100
#Parameters (2. SCF iterations)
scf_nmax 100
scf_thr 1e-8
#Parameters (3. Solve KS equation)
nbands 26
ks_solver cg
#Parameters (4.Smearing)
smearing_method gauss
smearing_sigma 0.01
#Parameters (5.Mixing)
mixing_type broyden
mixing_beta 0.7
mixing_gg0 0平面波(pw)作为一种可以用于描述周期性边界条件下的电子波函数和电荷密度基矢量,它们都是正交的,且可以通过一个 ecutwfc 参数来控制基矢量的个数,ecut 其实代表了每一个平面波所对应的动能,如果 ecut 取得越大,则基矢量可以描述震荡得越剧烈的物理量(例如氧原子的 2p 轨道),那么计算结果就会越精确,但同时所带来的机时成本消耗也越大。因此我们采用 pw 基组做真正计算前,需要对 ecut 进行测试来获得一个足够准确且效率也高的取值。
保持前文用于计算的基组为"pw",进行 ecut 的收敛性测试,ecut 取值范围为:20~100 Ry。这个例子在 4 进程下启动 MPI 进行并行计算,一般推荐总核数 (=线程数*进程数)取 2 的 n 次方或者 n 倍。一个较为粗糙、但基本不会造成计算资源浪费的选取 n 值原则是:体系有多少个原子,不要用超过这个原子个数太多的总核数进行并行计算。例如,体系如果有 16 个原子,不要用远大于 16 的总核数进行计算,一般取 16 的总核数或者更少就够了,具体需要测试。
mpirun -n 4 abacus
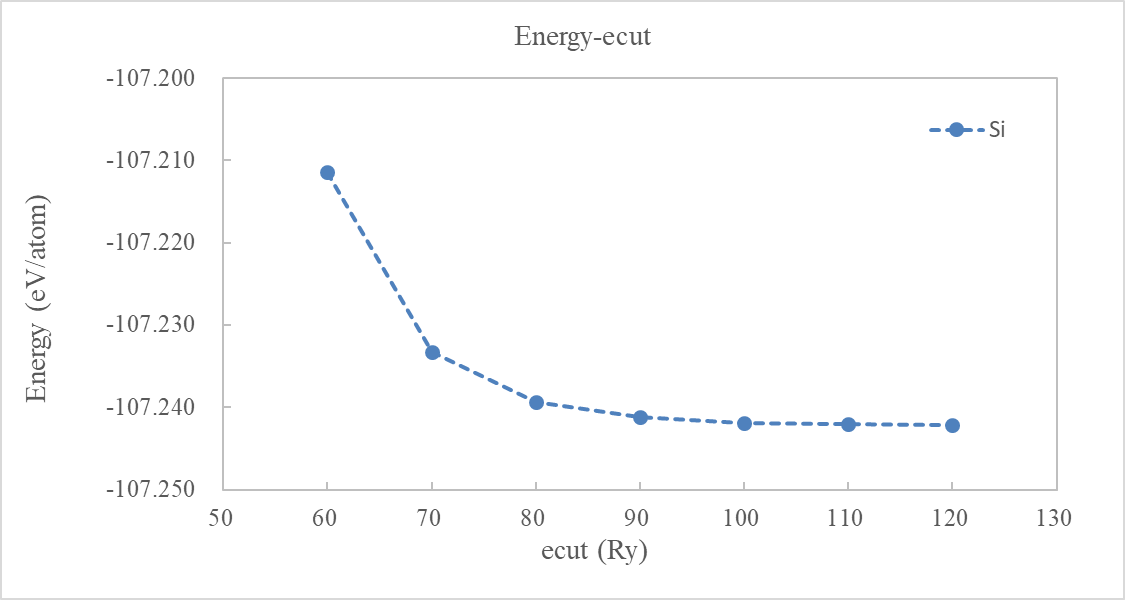
计算完成后,建议用脚本提取 ecut 数值下OUT.Si文件夹里running_scf.log文件中收敛的系统总能量,计算单原子能量并绘制其随着 ecut 的变化曲线。如图 2,可以看见随 ecut 增大,系统总能量趋于收敛。在 ecut=60 Ry 时,认为总能量收敛(与 ecut=50 Ry 的能量差小于 1 meV/atom)。
对具有周期性的体系进行计算时,DFT 计算实际会对第一布里渊区中不同离散化$k$点的单独进行计算(例如每个$k$点都会单独求解一次依赖于该$k$点的 Kohn-Sham 方程),并将计算结果进行积分。$k$点越多,则需要求的离散积分就越多,也将越近似连续积分的结果,但计算资源也会增加。因此,pw 和 LCAO 都有必要对 "需要多少$k$点" 进行测试并得出合适的$k$点选取方案。
金刚石 Si 的$k$点测试结果。同样在 4 进程下进行并行计算。
mpirun -n 4 abacus
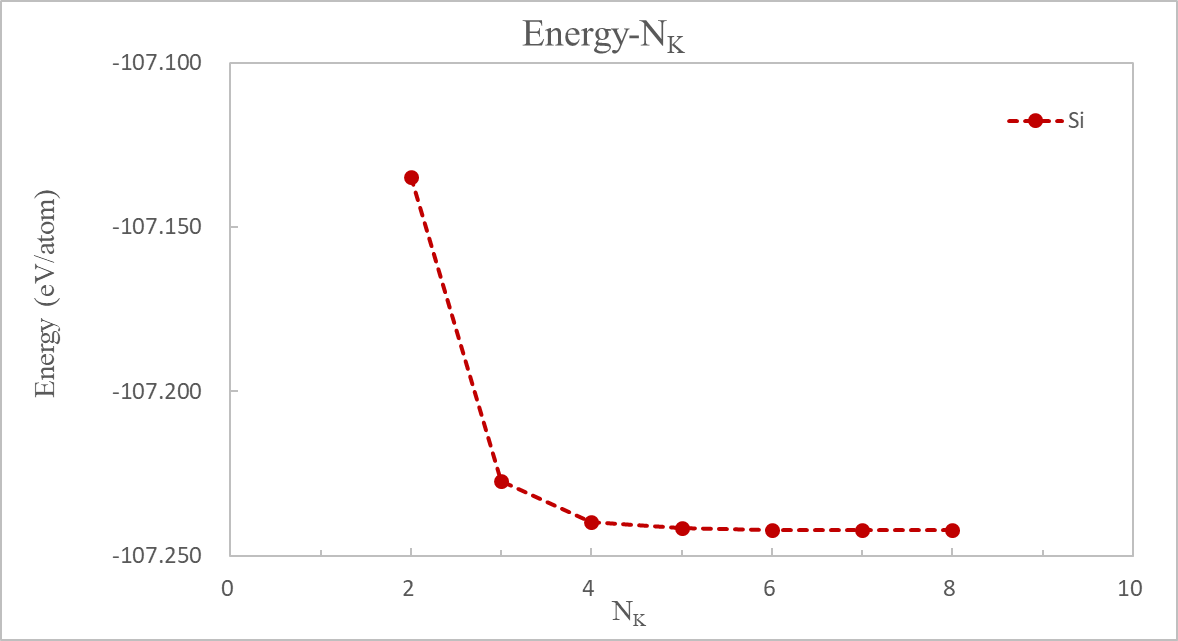
计算完成后,提取各 K 点下OUT.Si文件夹里running_scf.log文件中收敛的系统总能量和计算时间,绘制这两个参数随 K 点的变化曲线。此处,$N_k$表示各方向上的$k$点取值个数,"$Energy-N_k$" 图中采用单原子的能量。
分析图 3 和图 4 可知,在$N_k=6$时,结果已收敛,能量不会再发生较大的变化(与$N_k=5$的能量差小于 1 meV/atom),而此时花费时间 132 秒,相比于更大的$N_k$点较低。因此选择 6×6×6 的$k$点可以同时获得准确的结果和高的计算效率。
体系里平均单个 Si 原子能量 (in eV/atom) 随 K 点变化:
计算时间随 K 点变化:
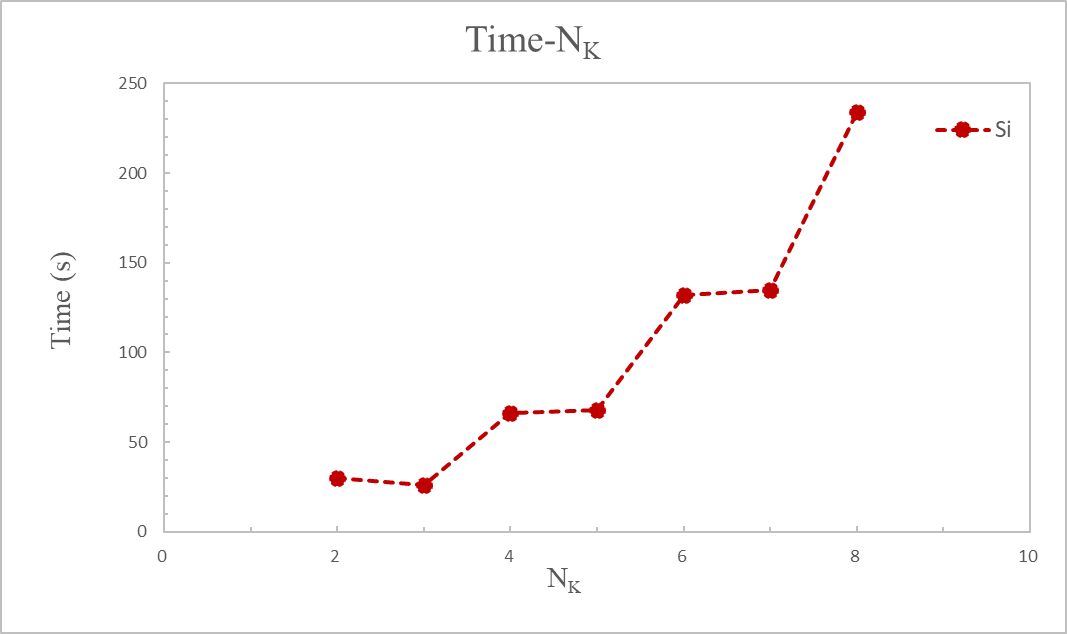
提取出每个计算中实际使用到$k$点个数,如表一所示,实际使用$k$点数相比总$k$点数有较明显的简化。如前所述,INPUT 文件中我们设置了参数 symmetry 取 "1",因此在实际计算中利用了晶胞结构中的对称性,即倒易空间的积分没有使用所有的$k$点计算,而是巧妙的利用了对称性得到其它$k$点的信息,简化了布里渊区中的计算工作量。这也解释了图 4 中明明总$k$点数不同,计算时间却十分接近的原因。
表一、$N_k$点数,总$k$点数,实际使用$k$点数:
| 2 | 3 | 4 | 5 | 6 | 7 | 8 | |
|---|---|---|---|---|---|---|---|
| 总$k$点个数 | 8 | 27 | 64 | 125 | 216 | 343 | 512 |
| 实际计算$k$点数(开了对称性) | 4 | 4 | 10 | 10 | 20 | 20 | 35 |